

The Hidden Bottleneck in Chemical Analysis

Chromatography–mass spectrometry is one of the most versatile tools in chemical analysis, capable of detecting a wide range of chemicals. Modern instruments like GC-MS and LC-MS generate a surprisingly large amount of data; up to hundreds of megabytes from a single run. But while the hardware has advanced rapidly, the software used to process and interpret this data hasn’t kept pace. Analysts still spend hours manually checking peaks, spectra, and library hits to ensure validity, slowing down workflows and limiting discovery.
Vendor software often relies on rigid, decades-old algorithms, making it hard to adapt to today’s needs. The result is a bottleneck: slower turnaround, higher error rates, and missed opportunities to detect compounds beyond standard libraries.

What We Do
That’s why we take a software-first approach to chemical identification, improving the reliability and turn around time of GC-MS and LC-MS/MS analysis not with fancier instrumentation, but with tailored, in-house software built on cutting-edge algorithmic methods. These methods are grounded in PhD research conducted at the University of Western Australia, combining deep domain knowledge in analytical chemistry with advances in signal processing, machine learning, and optimization methods.
Our software enables:
Faster Turnaround Times
Unlock new commercial opportunities for high-throughput testing and broader service offerings
Higher Identification Confidence
Reduce liability and improve reproducibility in regulated or risk-sensitive environments
Semi-automated Data Analysis
Allow your analysts to focus on interpreting results, not manually inspecting ion currents or mass peaks
Our Work
Current Projects
Intelligent Peak Deconvolution
Our system leverages the entire 2D mass chromatogram, applying advanced model fitting and clustering techniques to separate overlapping peaks with minimal manual intervention.
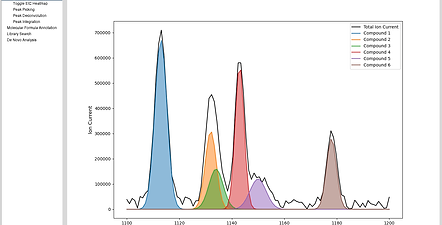
Improved Detection Sensitivity
Manual baseline subtraction is slow, subjective, and often inconsistent. Our system automates this process, detecting true mass peaks without analyst intervention. By eliminating noise and preserving peak integrity, we deliver more accurate mass measurements and highly reproducible spectra, enhancing sensitivity and freeing scientists to focus on the results.

Automated Molecular Formula Identification
Our system uses the cutting-edge Parent Subformula Graph method to automatically assign molecular formulae to mass peaks, making it much easier for analysts to reason chemically with mass spectral data.

Beyond Library Search
Our system combines standard library matching with structural database search and novel algorithms designed to infer molecular identity from spectral patterns. This enables computer-assisted identification even when a compound is absent from existing libraries, expanding the range of analytes that can be confidently detected.
